
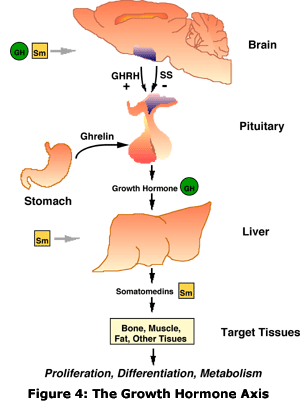
Os sinais e sintomas do hipopituitarismo variam de acordo com os hormônios afetados e a causa primária da anormalidade. O diagnóstico é feito através de exames de sangue, mas geralmente são necessários exames específicos e outras vistorias para que sejam identificadas as causas primárias e o tratamento a ser adotado. A maioria dos hormônios controlados pelas secreções pituitárias pode ter repostas por comprimidos ou injeções. O hipopituitarismo é uma doença rara, mas pode ser precisamente pré-diagnosticada em pessoas que já tiveram lesões cerebrais traumáticas. A primeira descrição da doença foi feita em 1914, pelo médico alemão Dr. Morris Simmonds. As malformações não são comuns, podem estar ligadas a defeitos ao nascer através de mutações genéticas. Ao nascimento, crianças com déficit de IGF-1 tem tamanho normal, mas aquelas com déficit ou insensibilidade congênita intensa ao GH são pequenas. Anormalidades anatômicas da região hipotálamo-hipofisária podem ser observadas por imagens obtidas pela ressonância magnética, incluindo disgenesia da haste hipotálamo-hipofisária, localização ectópica da hipófise posterior e volume diminuído da hipófise anterior. Déficit de GH ocorre com grande frequência em partos pélvicos e cesarianos. Sob o aspecto clínico, um desvio do normal na curva de crescimento deve sempre ser considerado patológico no período que compreende dos 2 anos de idade até o início da puberdade. As malformações congênitas envolvendo o hipotálamo levam com frequência ao hipopituitarismo. A síndrome da displasia septi-óptica na sua forma completa apresenta hipoplasia ou ausência do quiasma ótico, agenesia ou hipoplasia do septo pelúcido ou corpo caloso e insuficiência hipotalâmica. Pode ocorrer déficit isolado de GH ou associado à deficiência de TSH, ACTH, LH, e FSH. Alguns pacientes apresentam na ressonância magnética interrupção ou hipoplasia da haste hipotálamo-hipofisária e ectopia da neurohipófise com hipoplasia ou aplasia da hipófise anterior.
Déficit isolado de GH ou deficiências múltiplas de secreções da hipófise anterior podem ocorrer após trauma craniano, processos infecciosos, e ainda por irradiação da cabeça como indicação terapêutica, atingindo o hipotálamo ou a hipófise, entre outras patologias. Um grupo de crianças com baixa estatura e baixa velocidade de crescimento apresenta resposta normal de GH a testes provocativos, mas com diminuição de IGF-1 (fator de crescimento insulina símile–1) e diminuição de picos de GH quando se faz amostragem frequente durante períodos de 12 a 24 horas, preenchendo o critério de disfunção neurossecretória de GH. A síndrome de Prader-Willi, geneticamente determinada (deleção funcional do alelo paterno dentro do cromossomo 15q 11-13), caracteriza-se por hipotonia neonatal com subsequente diminuição de força e massa muscular, além de baixa estatura ao nascimento, que se acentuam no período pós-natal, associada à criptorquidismo e hipogonadismo hipogonadotrófico que podem persistir até a vida adulta, ocasião em que esses pacientes tornam-se muito obesos. A causa mais provável da baixa estatura é a produção DGH com consequente baixa de IGF-1. O grande problema é que as mal formações em geral são complexas, muitas vezes silenciosa e não aparente, entretanto, quando aparecem equivalem à ponta de um Iceberg onde 1/8 estará aparecendo sequencialmente e 7/8 infelizmente não é bem clara, outro sim, quanto mais precoce for feita a prevenção ou correção quando possível, mais eficiente é a solução.
GROW CHILD, INFANTILE AND YOUTH: THE BIRTH DEFECTS INVOLVING THE HYPOTHALAMUS OFTEN LEAD TO HYPOPITUITARISM.
GROWING UP: HYPOPITUITARISM IS A DISEASE CHARACTERIZED BY REDUCED ENDOCRINE SECRETION OF ONE OR MORE OF EIGHT HORMONES NORMALLY PRODUCED BY PITUITARY GLAND (PITUITARY), IN THE BASE OF THE BRAIN. IF THERE'S DECLINE OF MOST OF PITUITARY HORMONES, IS USED THE TERM PANHYPOPITUITARISM. PHYSIOLOGY-ENDOCRINOLOGY-NEUROENDOCRINOLOGY-GENETICS-ENDOCRINE-PEDIATRICS (SUBDIVISION OF ENDOCRINOLOGY): DR. JOÃO SANTOS CAIO JR. ET DRA. HENRIQUETA VERLANGIERI CAIO.

The signs and symptoms of Hypopituitarism vary with the affected hormones and the primary cause of the abnormality. Diagnosis is made through blood tests, but specific tests are usually required and other surveys for which the root causes are identified and the treatment to be adopted. Most hormones controlled by the pituitary secretions can be replaced by tablets or injections. Hypopituitarism is a rare disease, but it can be accurately pre-diagnosed in people who have had traumatic brain injuries. The first description of the disease was made in 1914 by German physician Dr. Morris Simmonds. The malformations are not common, can be linked to birth defects through genetic mutations. At birth, the children with deficits of IGF-1 has normal size, but those with deficits or severe congenital GH insensitivity are small. Anatomical abnormality of the hypothalamic-pituitary region can be observed by images obtained by MRI, including dysgenesis of the hypothalamic-pituitary stalk, ectopic posterior pituitary and location of anterior pituitary volume decreased. Deficit of GH - growth hormone occurs very frequently in pelvic and cesarean sections. From the clinical aspect, a deviation from normal in the growth curve should always be considered pathological in the period that includes two years of age and the onset of puberty. Congenital malformations involving the hypothalamus often lead to hypopituitarism. The syndrome of septic-optic dysplasia in its complete form shows hypoplasia or absence of the optic chiasm, agenesis or hypoplasia of the septum pellucid or corpus callosum and hypothalamic failure. Growth hormone or associated Hypopituitarism with TSH, ACTH, LH deficiency, and FSH - isolated GH deficit may occur. Some patients present on MRI interruption or hypoplasia of the hypothalamic-pituitary stalk and ectopic of the neurohypophysis with hypoplasia or aplasias of the anterior pituitary.

Isolated DGH or multiple anterior pituitary secretions may occur after head trauma, infectious processes, and also by irradiation of the head as therapeutic indication, reaching the hypothalamus or pituitary gland, among other pathologies. A group of children with short stature and low growth rate have normal GH response but with reduced IGF-1 (growth factor insulin simile – 1) and decreased GH peaks - hormone growth when making frequent sampling during periods of 12 to 24 hours, filling the criterion neurosecretory dysfunction GH. The Prader-Willi syndrome, genetically determined (functional deletion of the paternal allele in the chromosome 15q 11-13), characterized by neonatal hypotonia with subsequent loss of strength and muscle mass, and short stature at birth, which increases the postnatal period, associated with cryptorchidism and hypogonadism that can persist into adulthood, at which time these patients become very obese. The most likely cause of short stature is DGH production with consequent lower IGF-1. The big problem is that the formations evil in general are complex, often silent and not apparent, however appear equivalent when the tip of an iceberg where 1/8 and 7/8 will be appearing sequentially unfortunately is not very clear, but the other, as early prevention or correction is made when possible, is the most efficient solution.
Dr. João Santos Caio Jr.
Endocrinologia – Neuroendocrinologista
CRM 20611
Dra. Henriqueta V. Caio
Endocrinologista – Medicina Interna
CRM 28930
Como saber mais:
1. A síndrome de má absorção e doenças gastrintestinais: doença celíaca e doença de Crohn devem ser consideradas no diagnóstico diferencial do baixo crescimento linear tendo em vista a má absorção de nutrientes que são necessários para eficiência metabólica na fase de crescimento de criança, infantil, juvenil e adolescente...
http://hormoniocrescimentoadultos.blogspot.com
2. Anticorpos antigliadina, autoanticorpo antiendomísio, dos autoanticorpos antireticulina, antiendomísio e antitransglutaminase tecidual devem ser pesquisados para afastar doença celíaca...
http://longevidadefutura.blogspot.com
3. O diagnóstico acurado da doença celíaca é muito importante embora, muitas vezes, desafiador...
http://imcobesidade.blogspot.com
AUTORIZADO O USO DOS DIREITOS AUTORAIS COM CITAÇÃO
DOS AUTORES PROSPECTIVOS ET REFERÊNCIA BIBLIOGRÁFICA.
Referências Bibliográficas:
Caio Jr, João Santos, Dr.; Endocrinologista, Neuroendocrinologista, Caio,H. V., Dra. Endocrinologista, Medicina Interna – Van Der Häägen Brazil, São Paulo, Brasil; Wilkins L. The diagnosis and treatment of endocrine disorders in childhood and adolescence. Springfield, IL: Charles C Thomas Publisher, 1965; Greulich WW, Pyle SI. Radiographic atlas of skeletal development of the hand-wrist, 2nd ed. Palo Alto, CA: Stanford University Press, 1959; Laron Z. The hypothalamus and the pituitary gland. In: Hubble D, ed. Pediatric endocrinology. Oxford: Blackwell Scientific, 1969:35; Rimoin DL, Schimke RN. Genetic disorders of the endocrine glands. St. Louis: Mosby, 1971:11; Lieblich JM, Rogol AD, White BJ, Rosen SW. Syndrome of anosmia with hypogonadotropic hypogonadism (Kallmann syndrome). Am J Med 1982; 73:506; Triulzi F, Scotti G, diNatale B, et al. Evidence of a congenital midline brain anomaly in pituitary dwarfs: a magnetic resonance imaging study in 101 patients. Pediatrics 1994; 93:409; Lieblich JM, Rosen SW, Guyda H, et al. The syndrome of basal encephalocele and hypothalamic-pituitary dysfunction. Ann Intern Med 1978; 89:910; Procter AM, Phillips JA III, Cooper DN. The molecular genetics of growth hormone deficiency. Hum Genet 1998; 103:255; Rimoin DL. Hereditary forms of growth hormone deficiency and resistance. Birth Defects 1976; 12:15; Phillips JA III. The growth hormone (hGH) gene and human disease. In: Banberry Report 14: Recombinant DNA applications to human disease. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory, 1983; 305; Rogol AD, Blizzard RM, Foley TP Jr, et al. Growth hormone releasing hormone and growth hormone: genetic studies in familial growth hormone deficiency. Pediatr Res 1985; 19:489; Lovinger RD, Kaplan SL, Grumbach MM. Congenital hypopituitarism associated with neonatal hypoglycemia and microphallus: four cases secondary to hypothalamic hormone deficiencies. J Pediatr 1975; 87:1171; Paja M, Lucas T, Garcia-Uria J, et al. Hypothalamic-pituitary dysfunction in patients with craniopharyngioma. Clin Endocrinol 1995; 42:467; Burr IM, Slonim AE, Danish RK, et al. Diencephalic syndrome revisited. J Pediatr 1976; 88:439; Tanabe M, Watanabe T, Hori T: Von Recklinghausen’s disease with diencephalic syndrome in an adult: case report. J Neurosurg 1994; 80:556.